
认知是大脑皮层复杂高级功能的反映,任何直接或间接导致大脑皮层结构和功能慢性损伤的因素均可通过不同机制引起认知障碍,现将其归纳如下:
(1)神经递质及其受体异常:大多数神经元之间的信息传递是通过神经递质(neurotransmitter)及其相应的受体完成的。这些神经递质或受体异常改变均可导致不同类型和不同程度的认知异常。
1)多巴胺(dopamine):多巴胺是以酪氨酸为底物,在酪氨酸羟化酶(tyrosine hydroxylase)和多巴脱羧酶(dopamine decarboxylase)的作用下合成的。研究发现:脑中多巴胺含量显著降低时可导致动物智能减退、行为情感异常、言语错乱等高级神经活动障碍。例如,在帕金森病(Parkinson disease,PD)患者黑质多巴胺能神经元减少,酪氨酸羟化酶和多巴脱羧酶活性及纹状体多巴胺递质含量明显卞降。此外,在动物实验中发现多巴胺过多也可导致动物认知功能的异常改变。多巴胺受体有D1和D2受体两大家族,精神分裂症患者与大脑额叶皮层的D1受体功能低下和皮层下结构D2受体功能亢进双重因素有关,因此有人提出用D1激动和D2阻断治疗精神分裂症的新概念。
2)去甲肾上腺素(nonepinephrine):去甲肾上腺素是最早被发现的单胺类神经递质,是多巴胺经β羟化酶作用生成的产物。在脑内,去甲肾上腺素通过α1、α2和β受体发挥调节作用。在突触前,α2受体通过Gi蛋白介导,减少cAMP的生成和cAMP依赖性蛋白激酶的活性,减少蛋白激酶对N-型Ca2+通道的磷酸化,以至Ca2+通道关闭,Ca2+内流减少,从而对去甲肾上腺素的释放起抑制作甩(负反馈调节);α2受体激动还可抑制在警醒状态下的蓝斑神经元的放电增加;在突触后,α1受体激动可引起K+通道开放,K+外流增加,神经元倾向超极化而产生抑制效应。而α1受体激活则使K+通道功能降低,K+外流减少,神经元去极化产生兴奋效应。一般认为,脑中α2受体激动与维持正常的认知功能有关,而α1受体持续、过度激活可致认知异常。在正常警醒状态时,脑细胞含适量去甲肾上腺素,α2受体功能占优势,维持正常的认知功能。在应激状态下产生大量去甲肾肾上腺素,α1受体功能占优势;这可能是个体长期处于应激状态更易出现认知障碍的机制之一。
3)乙酰胆碱(aeetylcholine):乙酰胆碱由乙酰辅酶A和胆碱在胆碱乙酰转移酶的作用下生成。神经细胞合成并释放的乙酰胆碱通过M-受体(M-AchR,毒蕈碱受体)和N-受体(N-AchR,烟碱受体)发挥调节作用,M-AchR是G-蛋白耦联受体,N-AchR是配体门控离子通道受体。脑内的胆碱能神经元被分为两类,即局部环路神经元和投射神经元,自Meynert基底核发出的胆碱能纤维投射至皮层的额叶、顶叶、颞叶和视皮层,此通路与学习记忆功能密切相关。阿尔茨海默病(Alzheimer's disease,AD)患者在早期便有Meynert基底区胆碱能神经元减少,导致皮层胆碱乙酰转移酶活性和乙酰胆碱含量显著降低,是AD患者记忆障碍的重要机制之一;精神分裂症者认知障碍的程度与皮层胆碱乙酰转移酶活性呈负相关;给AD和精神分裂症患者使用胆碱酯酶抑制剂或M受体激动剂可改善其记忆缺损。
4)谷氨酸(glutamate):在脑内,氨基酸类递质含量最高,其中,谷氨酸在人大脑皮层中的含量约为9-11μmol/g,比乙酰胆碱或单胺类递质的含量高103数量级,比神经肽的含量高106数量级。谷氨酸是不能透过血脑屏障的非必需氨基酸,脑内的谷氨酸可分别由谷氨酰胺在谷氨酰胺酶的作用下水解或α-酮戊二酸在其转氨酶的作用下生成。谷氨酸藉N-甲基-D-门冬氨酸(N-methyl-D-aspartate,NMDA)和非NMDA受体起作用。NMDA受体是配体门控的离子通道型受体;非NMDA受体主要指海人藻酸(kainate,KA)和α-氨基-3-羟基-5-甲基-4-异恶唑-丙酸(α-mino-3-hydroxy-5-methy-4-isoxa-zolep-propionate,AMPA)是Na+-K+通透性离子通道型受体。纹状体的谷氨酸神经纤维抑制丘脑向大脑皮层发出感觉冲动,当谷氨酸能神经低下时,这种冲动发出增多,大脑皮质单胺活性增强,引起相应的认知功能异常。由于谷氨酸是哺乳动物脑内最重要的兴奋性神经递质,故当谷氨酸含量异常增高时,可引起“兴奋性毒性”损伤(见后述)。
(2)神经肽异常:神经肽(neuropeptide)是生物体内的一类生物活性多肽,主要分布于神经组织。在脑内,神经肽与神经递质(neurotransmitter)常常共存于同一神经细胞,但神经肽与经典神经递质有诸多不同:神经肽比神经递质分子量大,在脑组织中含量低;神经肽由无活性的前体蛋白加工而成,而神经递质可在胞体或神经末梢直接合成;神经肽释放后主要经酶解而失活,神经递质则主要通过神经末梢重吸收反复利用;神经肽的调节缓慢而持久,神经递质的调节快速而精确等。神经肽的异常与认知障碍密切相关。有人报道PD患者脑苍白球和黑质中P物质水平下降30%-40%,在黑质中胆囊收缩素(cholecystokinin,CCK)下降30%,在丘脑下部和海马区神经降压肽(neurotensin,NT)含量也下降。血管加压素(vasopressin,VP),血管活性肠肽(vasoac-tire intestinal peptide,VIP)及其受体含量减少与记忆力减退相关,给脑外伤、慢性乙醇中毒及AD病人用VP可改善其记忆力减退。促甲状腺素释放激素(thyrotropin releasing hormone,TRH)是第一个从丘脑下部分离出来的三肽激素,TRH可引起行为改变,如兴奋、精神欣快及情绪暴躁等。TRH既可以作为一种神经激素通过受体调节其他递质起作用,又可以作为一种神经递质直接起作用。腺垂体分泌的促肾上腺激素释放激素(adrenocorticotropic hormone,ACTH)是一39肽激素,其水平改变影响动物的学习记忆、动机行为等。ACTH影响动物学习和行为的关键分子区域是其分子中第4~10位氨基酸残基,该片断能提高大鼠的注意力和记忆力,同时减轻动物的焦虑行为。多发性硬化(multiple sclerosis,MS)患者丘脑下部-垂体一肾上腺皮质(hypothalamus-pynear-adrenocorticode,HPA)轴功能紊乱与其反应迟钝、智能低下、重复语言等认知功能障碍显著相关。根据绝经期女性AD的发病率高于男性,且经绝后接受雌激素替代疗法者的患病率降低,有人提出性激素代谢紊乱也可能参与认知障碍的发病过程。
(3)神经营养因子缺乏:神经元和胶质细胞可合成、分泌大量的神经营养因子,如神经生长因子(neurogrowth factor,NGF)、睫状神经营养因子(ciliary neurotrophic factor,CNTF)、脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)和胶质源性神经营养因子(glia-derived neu-rotrophic factor,GDNF)等。这些神经营养因子对神经元的存活和神经元突起的生长具有重要作用。已发现在多种神经退行性疾病中均有神经营养因子含量的改变,例如,在PD患者黑质NGF、BDNF和GDNF的含量明显降低,离体和在体实验均证明BDNF、GDNF和CNTF对吡啶类衍生物1-甲基4一苯基l,2,3,6一四氢吡啶(MPTP)造成的多巴胺能神经元损伤具有很强的保护作用。
脑组织中蛋白质异常聚集可见于一大类脑神经细胞退行性变性疾病中,如AD、PD、亨廷顿病(Huntington disease,HD)、海绵状脑病(Creutzfeldt Jokob disease,CJD)等。蛋白质的异常聚积与基因变异、蛋白质合成后的异常修饰、脑组织慢病毒感染、脑老化和环境毒素中毒等多种因素有关。
(1)基因异常:已发现多种基因异常参与神经细胞的退行性变性。例如,在PD患者有ot-synuclein,parkin和park3基因突变,a-synuclein基因第209位的核苷酸发生了G-A错义突变,使其蛋白质第53位的丙氨酸(Ala)变成了苏氨酸(Thr),变异的蛋白质是PD患者神经细胞胞浆中特征性嗜酸性包涵体,即路易(Lewy)小体的重要成分;已发现有30多种不同parkin基因缺失和点突变与早发性PD有关,改变的parkin蛋白可导致依赖泛素的蛋白降解过程异常,促使parkin蛋白聚集。在AD患者,已发现5个相关基因突变,所编码的蛋白质依次为淀粉样前体蛋白(amy-loid precursor protein,APP)、早老蛋白-1(presenilin-1,PS-1)、PS-2、载脂蛋白E(apolipoprotein E,apoE)和α2-巨球蛋白(α2-macro谷氨酸bumin)。其中,APP、PS基因突变和ApoE基因多态性可导致APP异常降解,产生大量B淀粉样多肽(AB),过量产生的Ap不断在神经细胞间聚集形成老年斑,同时可导致过氧化损伤(损伤生物膜、破坏细胞内钙离子稳态、抑制星形胶质细胞、使一些关键酶失活)、炎症反应和神经细胞死亡。
(2)蛋白质合成后的异常修饰:正常时,蛋白质合成后的不同加工修饰赋予蛋白质不同的结构和功能,是蛋白质结构和功能多样性的基础。蛋白质的异常修饰导致其结构异常、功能降低或丧失。在AD患者,发现细胞骨架蛋白tau被异常磷酸化(phosphorylation)、异常糖基化(glycosylmion,酶促反应)、异常糖化(glycmion,非酶促反应)和异常泛素化(ubiquitilation)修饰,异常修饰的tau蛋白沉积在神经细胞中形成神经原纤维缠结。关于tau蛋白异常糖基化、异常糖化和异常泛素化的机制尚不清楚,目前认为AD患者tau蛋白被异常磷酸化可能与蛋白磷酸酯酶(proteinphosphatase)和蛋白激酶(protein kinase)调节失衡有关。蛋白磷酸酯酶催化蛋白质去磷酸化,AD患者脑中蛋白磷酸酯酶的活性明显降低,使tau蛋白去磷酸化减弱,导致AD患者脑中tau蛋白异常过度磷酸化。蛋白激酶催化蛋白质磷酸化,在AD患者,大脑颞叶皮层多种蛋白激酶的表达量或活性比对照者显著增强。上述磷酸化系统失衡导致tau蛋白异常过度磷酸化,异常修饰的tau在神经细胞内聚集是AD患者神经细胞退化的重要机制。
(3)脑组织慢病毒感染:最常见的由慢病毒感染引起的人类中枢性疾病为CJD,是由一种具传染性的朊蛋白(prion protein,PrP)所致。这种PrP类似于病毒可传播疾病,但与已知病毒不同是,它没有任何可检测到的核酸序列。人类PrP蛋白有两种异构体,分别是存在于正常细胞的PrP(PrPc)和引起朊蛋白病的PrPsc(PrP scrapie)。两种异构体的序列并无差别,但蛋白质的空间构型不同。PrPc是一种细胞内膜结合蛋白,PrPsc不仅存在于细胞内膜,还存在于朊蛋白病患者神经细胞外的淀粉样蛋白纤丝和斑块中;prpsc可促进PrPc转化为PrPsc。在人体内,PrPsc的增殖是通过一分子PrPc与一分子PrPsc结合形成杂二聚体,此二聚体再转化成两分子PrPsc,PrPsc便依此呈指数增殖。有朊蛋白基因突变时,细胞中的PrPc。更易从α-螺旋转变成β-片层,此时更容易与PrPsc结合,导致PrPsc增殖和聚集。
神经元能量储备极少,对缺血、缺氧非常敏感,完全缺血5分钟即可导致神经元死亡。脑缺血造成大脑皮层损伤是引起不同类型认知障碍的常见原因。统计资料表明:脑卒中患者在发病后出现痴呆的危险性较同龄对照组明显增高;有脑卒中史的老年群体的认知水平亦低于无卒中史的同龄老人。脑细胞缺血引起认知异常的机制可能与下述因素有关。
(1)能量耗竭和酸中毒:在缺血、缺氧状态下,细胞的能量代谢转为无氧酵解。无氧酵解生成ATP的效率低,使细胞出现能量耗竭。无氧酵解引起脑组织缺血性乳酸酸中毒,细胞Na+-K+泵功能损伤,K+大量外溢,同时Na+、Cl-及Ca2+大量流人细胞内引起细胞损伤;缺血区乳酸堆积还可引起神经胶质和内皮细胞的水肿和坏死,加重缺血性损害。
(2)细胞内Ca2+超载:脑缺血时,神经细胞膜去极化,引起大量神经递质释放,兴奋性递质(如谷氨酸)的释放激活NMDA受体,使钙通道开放,Ca2+内流增加;如激活非NMDA受体,使Ca2+从内质网释放至细胞浆内;膜去极化本身也启动了电压依赖性钙通道,加重Ca2+内流。神经细胞Ca2+超载可通过下述机制导致细胞死亡:①Ca2+超载时,大量Ca2+沉积于线粒体,干扰氧化磷酸化,使能量产生障碍;②激活细胞内Ca2+依赖性酶类,其中Ca2+依赖的中性蛋白水解酶过度激活可使神经细胞骨架破坏;③激活磷脂酶A和磷脂酶C,使膜磷脂降解;产生大量游离脂肪酸,特别是花生四烯酸,后者在代谢过程中产生血栓素、白三烯,一方面通过生成大量自由基加重细胞损害;另一方面可激活血小板,促进微血栓形成,在缺血区增加梗死范围,加重脑损害;④脑缺血时,脑血管平滑肌,内皮细胞均有明显Ca2+超载,前者可致血管收缩、痉挛,血管阻力增加,延迟再灌流,使缺血半暗带内侧支循环不能形成,从而脑梗死灶扩大;后者可致内皮细胞收缩,内皮间隙扩大,血脑屏障通透性增高,产生血管源性脑水肿。
(3)自由基损伤:在急性脑缺血时,自由基产生和清除平衡状态受到破坏而引起脑损伤。其机制为:①缺血脑细胞能量衰竭,谷氨酸、天门冬氨酸(Asp)增多,此时电压依赖性钙通道和NM.DA受体操纵的钙通道开放,钙离子大量内流,使黄嘌呤脱氢酶转化为黄嘌呤氧化酶,后者催化次黄嘌呤氧化为黄嘌呤并同时产生氧自由基;钙离子大量内流还可激活磷脂酶A,造成血管内皮细胞和脑细胞的膜磷脂降解,花生四烯酸产生增加,后者代谢产生自由基;②缺血区脑细胞线粒体内钙离子增多,三羧酸循环发生障碍,不能为电子传递链的细胞色素氧化酶提供足够的电子将O2还原成H2O,从而生成氧自由基,并漏出线粒体;③急性脑缺血时,NO增多,NO能与氧自由基相互作用形成过氧亚硝基阴离子,后者又分解成羟自由基(OH-)和二氧化氮自由基(NO2-);④梗死灶内游离血红蛋白和铁离子与存在于细胞内的H202发生反应,产生OH-和氧自由基。儿茶酚胺等物质亦可发生氧化反应生成氧自由基。⑤缺血灶由于趋化因子增加,在血管内皮表面吸附大量中性粒细胞和血小板,前者通过细胞色素系统和黄嘌呤氧化酶系统产生O氧自由基和H202,后者通过血小板活化因子引起细胞内Ca2+浓度升高,促进自由基生成。
(4)兴奋性毒性:中枢神经系统中大部分神经递质是氨基酸类,包括谷氨酸、天冬氨酸、γ-氨基丁酸(GABA)和甘氨酸。其中,谷氨酸和天冬氨酸对神经元有极强的兴奋作用,故称为兴奋性氨基酸(excitatory amino acid,EAA),GABA和甘氨酸对神经元行使抑制作用,故称为抑制性氨基酸(inhibitory amino acid,IAA)。“兴奋性毒性(excitatory toxicity)”指脑缺血缺氧造成的能量代谢障碍直接抑制细胞质膜上Na+-K+-ATP酶活性,使胞外K+浓度显著增高,神经元去极化,EAA在突触间隙大量释放,因而过度激活EAA受体,使突触后神经元过度兴奋并最终死亡的病理过程。EAA通过下述两种机制引起“兴奋性毒性”:一是AMPA受体和KA受体过度兴奋引起神经细胞急性渗透性肿胀,可在数小时内发生,以Na+内流,以及Cl-和H2O被动内流为特征;另一种是NMDA受体过度兴奋所介导的神经细胞迟发性损伤,可在数小时至数日发生,以持续的Ca2+内流为特征。
(5)炎症细胞因子损害:在脑缺血损害发生后,产生多种多效性细胞因子。在致炎细胞因子占主导地位时,加重脑缺血损害,在抗炎因子占主导时,对脑缺血产生保护作用。如白细胞介素-1β(IL-1β)和肿瘤坏死因子-α(TNF-α)加重脑缺血损害,转化生长因子β1(TGFβ1)对脑缺血有保护作用。此外,在缺血损伤的神经元释放的细胞因子激发下,缺血区吞噬细胞明显增加,吞噬细胞既能释放细胞因子刺激修复过程,又可释放神经毒素杀伤存活神经元。
对绝大多数50岁以后发病的典型散发性神经退行性疾病而言,环境和代谢毒素对脑的损害起主要作用,这些风险因素包括毒品、药物、酒精或重金属中毒等。各种慢性代谢性或中毒性脑病时,如心肺衰竭、慢性肝性脑病、慢性尿毒症性脑病、贫血、慢性电解质紊乱、维生素B缺乏、叶酸缺乏等,其主要表现为认知异常。
脑外伤对学习记忆和智力有不同程度的影响。轻度外伤者可不出现症状;中度外伤者可失去知觉;重度者可导致学习记忆严重障碍,乃至智力丧失。例如,一些“被打得昏头转向”的拳击手,脑反复损伤可出现构语障碍(口吃),心不在焉,好争辩,注意力涣散,近期记忆减退,步态僵硬、痉挛等。
认知功能一般随年龄增高(约60岁以后)而下降。研究发现,PD患者黑质多巴胺能神经元、酪氨酸羟化酶和多巴脱羧酶活力、纹状体多巴胺递质自30岁以后随年龄增长而逐年减少或降低。老年人脑巾血液供应减少,台成和分解代谢以及对毒素的清除能力均降低,这些都是造成老化脑神经细胞死亡,认知功能降低的主要因素。
心血管系统病变,如高血压、糖尿病、慢性阻塞性肺疾病等,可通过减少脑血液供应等机制,继发性降低大脑功能而引起认知障碍。处于亚临床阶段的心、脑血管疾病的高危人群,其认知测验的得分明显低于无任何亚临床特征的同龄老人,说明这些病变可能已经造成腑部的缺血、缺氧及脑功能损伤。此外,整体功能水平降低,如老年人听力下降使其与外界环境的接触以及对外界刺激的加工减少,也可降低老年人对外界环境的感知和认同;躯体功能,特别是操作性活动减少也可导致认知功能减退。有人发现,冠脉搭桥手术后的患者常出现短期记忆丧失和注意力下降,还有人认为,任何一种大的外科手术都可能导致大脑皮层功能的上述改变。
轻松、愉快、多彩的生活环境可促进实验动物大脑皮层的增长,使脑重量增加。相反,不良的心理、社会因素,如负性生活事件、处境困难、惊恐、抑郁等均可成为认知障碍的诱囡。近年来,利用电子计算机x线断层扫描(CT)与磁共振(MRI)对精神活动失调患者的脑成像研究发现,社会心理功能减退患者的有关脑区的皮层萎缩。用正电子发射扫描(PET)和单光子发射计算机断层扫描(SPECT)结合同位索示踪对脑局部脑血流(rCBF)和18氟一脱氧葡萄糖(FDG)或11碳-脱氧葡萄糖(CDG)代谢的研究证实,精神失常患者的有关脑区局部血流低灌注,葡萄糖利用率降低。用电子显微镜观察并经图像分析发现,精神分裂症患者的有关脑区神经细胞数目减少,细胞体积变小。
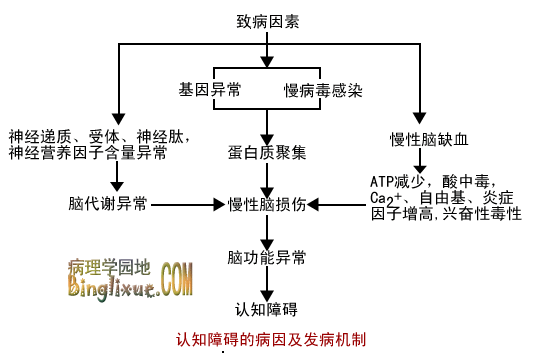
在诸多的人文因素中,受教育程度是报告最多、结果最恒定的影响认知的因素,认知测验的得分与受教育年限呈负相关。社会地位低下,经济生活状况较差与认知功能减退和痴呆的发生有一定关系。但在多因素分析中控制了年龄、性别、卒中史等较重要的因素后,社会经济凼素的影响一般不再显著。此外,女性认知功能损害的发生率高于男性,对各年龄组进行多因素分析的结果表明,这种差异与女性的受教育程度较低和慢性病患病率较高有关。图18-2简要概括认知障碍的病因和发病机制。
概念及认知的脑结构基础 | 主要表现形式 | 病因及发病机制 | 防治的病理生理基础