
在多细胞生物中,细胞与细胞之间的相互沟通除直接接触外,更主要的是通过内分泌、旁分泌和自分泌一些信息分子来进行协调。细胞通过位于胞膜或胞内的受体感受胞外信息分子的刺激,经复杂的细胞内信号转导系统的转换而影响其生物学功能,这一过程称为细细胞信号转导(cellular signal transduction),这是细胞对外界刺激做出应答反应的基本生物学方式。其中,水溶性信息分子如肽类激素、生长因子及某些脂溶性信息分子(如前列腺素)等,不能穿过细胞膜,需通过与膜表面的特殊受体相结合才能激活细胞内信息分子,经信号转导的级联反应将细胞外信息传递至胞浆或核内,调节靶细胞功能,这一过程称为跨膜信号转导(transmembrane signal transduction)。脂溶性信息分子如类固醇激素和甲状腺素等能穿过细胞膜,与位于胞浆或核内的受体结合,激活的受体作为转录因子,改变靶基因的转录活性,从而诱发细胞特定的应答反应。在病理状况下,由于细胞信号转导途径的单一或多个环节异常,可以导致细胞代谢及功能紊乱或生长发育异常。
G蛋白是指可与鸟嘌呤核苷酸可逆性结合的蛋白质家族,分为两类:①由α、β和γ亚单位组成的异三聚体,在膜受体与效应器之间的信号转导中起中介作用;②小分子G蛋白,为分子量21~28kD的小肽,只具有G蛋白α亚基的功能,在细胞内进行信号转导。目前发现的G蛋白偶联受体(G protein coupling receptors, GPCRs)已达300种以上,它们在结构上的共同特征是单一肽链7次穿越膜,构成7次跨膜受体。当受体被配体激活后,Gα上的GDP 为GTP所取代,这是G蛋白激活的关键步骤。此时G蛋白解离成GTP-Gα和Gβγ两部分,它们可分别与效应器作用,直接改变其功能,如离子通道的开闭;或通过产生第二信使影响细胞的反应。Gα上的GTP酶水解GTP,终止G蛋白介导的信号转导。此时,Gα与Gβγ又结合成无活性的三聚体。
(一) 腺苷酸环化酶途径(Adenylyl Cyclase Signal Transduction Pathway)
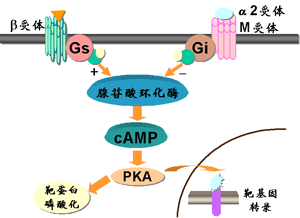
在腺苷酸环化酶(adenylyl cyclase,AC)信号转导途径中存在着两种作用相反的G蛋白,Gs与Gi。它们通过增加或抑制AC活性来调节细胞内cAMP浓度,进而影响细胞的功能。β肾上腺素受体、胰高血糖素受体等激活后经Gs增加AC活性,促进cAMP生成。而α2肾上腺素能受体、M2胆碱能受体及血管紧张素II受体等激活则与Gi偶联,经抑制AC活性减少cAMP的生成。cAMP可激活蛋白激酶A(protein kinase A,PKA),引起多种靶蛋白磷酸化,调节其功能。例如,肾上腺素引起肝细胞内cAMP增加,通过PKA促进磷酸化酶激酶活化,增加糖原分解。心肌β受体兴奋引起的cAMP增加经PKA促进心肌钙转运,提高心肌收缩力。进入核内的PKA可磷酸化转录因子CRE结合蛋白(cAMP response element binding protein, CREB),使其与DNA调控区的cAMP反应元件(cAMP response element, CRE)相结合,激活靶基因转录(图)。
(二)IP3、Ca2+-钙调蛋白激酶途径
α1肾上腺素能受体、内皮素受体、血管紧张素II受体等激活可与Gqα结合,激活细胞膜上的磷脂酶C(phospholipase C, PLC)β亚型,催化质膜磷脂酰肌醇二磷酸(phosphatidylinositol 4,5-diphosphate,PIP2)水解,生成三磷酸肌醇(1,4,5-inositol triphosphate, IP3)和甘油二酯(1,2-diacylglycerol,DG)。IP3促进肌浆网或内质网储存的Ca2+释放,Ca2+亦可作为第二信使启动多种细胞反应。例如,促进胰岛β细胞释放胰岛素;与心肌和骨骼肌的肌钙蛋白结合,触发肌肉收缩。Ca2+与钙调蛋白结合,激活Ca2+/钙调蛋白依赖性蛋白激酶活性,经磷酸化多种靶蛋白产生生物学作用。
(三)DG-蛋白激酶C途径
DG与Ca2+能协调促进蛋白激酶C(protein kinase C,PKC)活化。激活的PKC可促进细胞膜Na+/H+交换蛋白磷酸化,增加H+外流;PKC激活也可通过磷酸化转录因子AP-1、NF-KB 等,促进靶基因转录和细胞的增殖与肥大(图)。
(一)受体酪氨酸蛋白激酶途径(Receptor Tyrosine Protein Kinase Pathway)
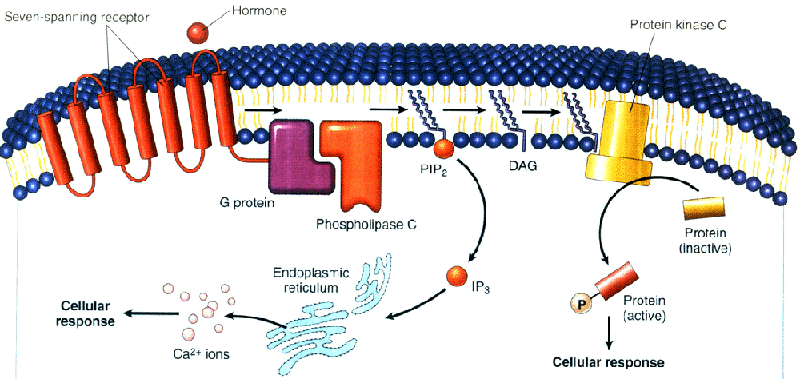
受体酪氨酸蛋白激酶(tyrosine protein kinase,TPK)是由50多种跨膜受体组成的超家族,其共同特征是受体胞内区含有TPK,配体则以生长因子为代表。表皮生长因子(epidermal growth factor, EGF)、血小板源生长因子(platelet-derived growth factor, PDGF)等与受体胞外区结合后,受体发生二聚化并催化胞内区酪氨酸残基自身磷酸化,进而活化TPK。磷酸化的酪氨酸可被一类含有SH2区(Src homology 2 domain)的蛋白质识别,通过级联反应向细胞内进行信号转导。由于大多数调节细胞增殖及分化的因子都通过这条途径发挥作用,故它与细胞增殖肥大和肿瘤发生的关系十分密切。
1.经Ras蛋白激活丝裂原活化蛋白激酶

丝裂原活化蛋白激酶(mitogen activited protein kinase,MAPK)家族是与细胞生长、分化、凋亡等密切相关的信号转导途径中的关键物质,可由多种方式激活。EGF、PDGF等生长因子与其受体结合并引起TPK激活后,细胞内含SH2区的生长因子受体连接蛋白Grb2与受体结合,将胞浆中具有鸟苷酸交换因子活性的Sos吸引至细胞膜,Sos 促进无活性Ras所结合的GDP为GTP 所置换,导致Ras活化。激活的Ras活化Raf(又称MAPK kinase kinase,MAPKKK),进而激活MEK(又称MAPK kinase,MAPKK),最终导致细胞外信号调节激酶(extracellular signal regulated kinase,ERK)激活(图)。激活的ERK可促进胞浆靶蛋白磷酸化或调节其他蛋白激酶的活性,如激活磷脂酶A2;激活调节蛋白质翻译的激酶等。更重要的是激活的ERK进入核内,促进多种转录因子磷酸化,如ERK促进血清反应因子(serum response factor, SRF)磷酸化,使其与含有血清反应元件(serum response element, SRE)的靶基因启动子相结合,增强转录活性。
2.经磷脂酶Cγ激活蛋白激酶C
受体TPK的磷酸化酪氨酸位点可为含有SH2区的PLCγ结合,导致PLCγ激活,水解PIP2生成IP3和DG,进而调节细胞的活动。
3.激活磷脂酰肌醇3激酶(phosphoinsitol 3' kinase,PI3K)
PI3K是由 p85调节亚单位和p110催化亚单位组成的异二聚体,因可催化磷脂酰肌醇3位的磷酸化而得名。PI3K的 p85与受体磷酸化的酪氨酸相结合,调节p110催化亚单位的活性,促进底物蛋白磷酸化,在细胞生长与代谢的调节中发挥重要作用。例如,PI3K可促进细胞由G1期进入S期;p110能与Ras-GTP结合,参与细胞生长的调节。
(二)非受体酪氨酸蛋白激酶信号转导途径
细胞因子如白介素(IL)、干扰素(INF)及红细胞生成素等的膜受体本身并无蛋白激酶活性,其信号转导是由非受体TPK介导的。非受体TPK的调节机制差异较大,现以INFγ为例,说明其信号转导途径。INFγ与受体结合并使受体发生二聚化后,受体的胞内近膜区可与胞浆内非受体TPK JAK激酶(janus kinase)结合并发生磷酸化,进而与信号转导和转录激活因子(signal transducers and activators of transcription,STAT)相结合。在JAK催化下,STAT中的酪氨酸磷酸化,并结合成STAT二聚体转移入核,与DNA启动子的活化序列结合,诱导靶基因的表达,促进多种蛋白质的合成,进而增强细胞抵御病毒感染的能力。
三、鸟苷酸环化酶信号转导途径
鸟苷酸环化酶(guanylyl cyclase,GC)信号转导途径存在于心血管系统和脑内,一氧化氮(nitric oxide,NO)激活胞浆可溶性GC,心钠素及脑钠素激活膜颗粒性GC,增加cGMP生成,再经激活蛋白激酶G(protein kinase G,PKG)磷酸化靶蛋白发挥生物学作用。
四、核受体(Nuclear receptor)及其信号转导途径
细胞内受体分布于胞浆或核内,本质上都是配体调控的转录因子,均在核内启动信号转导并影响基因转录,故统称为核受体(nuclear receptor)。按核受体的结构与功能可将其分为:①类固醇激素受体家族 包括糖皮质激素、盐皮质激素、性激素受体等。类固醇激素受体(除雌激素受体位于核内)位于胞浆,未与配体结合前与热休克蛋白(heat shock protein,HSP)结合存在,处于非活化状态。配体与受体的结合使HSP与受体解离,暴露DNA结合区。激活的受体二聚化并转移入核,与DNA上的激素反应元件(hormone response element,HRE)相结合或与其他转录因子相互作用,增强或抑制靶基因转录;②甲状腺素受体家族 包括甲状腺素、维生素D和维甲酸受体等。此类受体位于核内,不与HSP结合,多以同源或异源二聚体的形式与DNA或其他蛋白质结合,配体入核与受体结合后,激活受体并经HRE调节基因转录。
近年来的研究表明,不同信息分子、不同信号转导途径之间还存在交互调节(cross-talk),从而构成复杂的信号转导网络。例如,Giβγ可通过激活PLCβ,引起AC及PLC介导的信号转导途径之间的交互调节;某些GPCRs可通过促进非受体TPK磷酸化,增加Grb2和Sos结合,以Ras依赖形式激活ERK;或是经PKC激活Raf,以非Ras依赖形式激活ERK。在众多的信号转导途径中,无论任何环节出现异常,都可使相应的信号转导过程受阻,导致细胞的应答反应减弱、丧失或者反应过度,这些均可导致疾病的发生
疾病时的细胞信号转导异常可涉及受体、胞内信号转导分子及转录因子等多个环节。在某些疾病,可因细胞信号转导系统的某个环节原发性损伤引起疾病的发生;而细胞信号转导系统的改变亦可以继发于某种疾病或病理过程,其功能紊乱又促进了疾病的进一步发展。
因受体的数量、结构或调节功能变化,使之不能介导配体在靶细胞中应有的效应所引起的疾病称为受体病(receptor disease)。受体异常可以表现为受体下调(down regulation)或减敏(desensitization),前者指受体数量减少,后者指靶细胞对配体刺激的反应性减弱或消失。受体异常亦可表现为受体上调(up regulation)或增敏(hypersensitivity),使靶细胞对配体的刺激反应过度,二者均可导致细胞信号转导障碍,进而影响疾病的发生和发展。下表列出了部分与受体信号转导障碍相关的疾病,受体病按其病因可分为:
| 分类 | 累及的受体 | 主要临床特征 |
| 遗传性受体病 膜受体异常 家族性高胆固醇血症 |
LDL受体 |
血浆LDL升高,动脉粥样硬化 |
| 家族性肾性尿崩症 | ADH V2型受体 | 男性发病,多尿、口渴和多饮 |
| 视网膜色素变性 | 视紫质 | 进行性视力减退 |
| 遗传性色盲 | 视锥细胞视蛋白 | 色觉异常 |
| 严重联合免疫缺陷症 | IL-2受体γ链 | T细胞减少或缺失,反复感染 |
| II型糖尿病 | 胰岛素受体 | 高血糖,血浆胰岛素正常或升高 |
| 核受体异常 ccc 雄激素抵抗综合征 |
雄激素受体 |
不育症,睾丸女性化 |
| 维生素D抵抗性佝偻病 | 维生素D受体 | 佝偻病性骨损害,秃发,继发性甲状旁腺素增高 |
| 甲状腺素抵抗综合征 | β甲状腺素受体 | 甲状腺功能减退,生长迟缓 |
| 雌激素抵抗综合征 | 雌激素受体 | 骨质疏松,不孕症 |
| 糖皮质激素抵抗综合征 | 糖皮质激素受体 | 多毛症,性早熟,低肾素性高血压 |
| 自身免疫性受体病 cccccccccccccccc 重症肌无力 |
Ach受体 |
活动后肌无力 |
| 自身免疫性甲状腺病 | 刺激性TSH受体 抑制性TSH受体 |
甲亢和甲状腺肿大 甲状腺功能减退 |
| II型糖尿病 | 胰岛素受体 | 高血糖,血浆胰岛素正常或升高 |
| 艾迪生病 | ACTH受体 | 色素沉着,乏力,血压低 |
| 继发性受体异常 ccccccccccccccc 心力衰竭 |
肾上腺素能受体 |
心肌收缩力降低 |
| c帕金森病 | 多巴胺受体 | 肌张力增高或强直僵硬 |
| 肥胖 | 胰岛素受体 | 血糖升高 |
| 肿瘤 | 生长因子受体 | 细胞过度增殖 |
(一) 遗传性受体病
由于编码受体的基因突变使受体缺失、减少或结构异常而引起的遗传性疾病。
1. 家族性高胆固醇血症(familial hypercholesterolemia, FH)
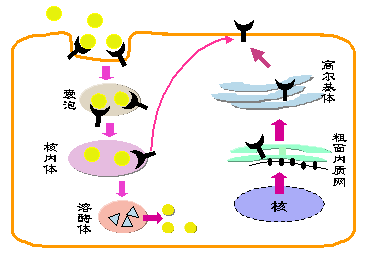
在肝细胞及肝外组织的细胞膜表面广泛存在着低密度脂蛋白(LDL)受体,它能与血浆中富含胆固醇的LDL颗粒相结合,并经受体介导的内吞作用进入细胞。在细胞内受体与LDL解离,再回到细胞膜,而LDL则在溶酶体内降解并释放出胆固醇,供给细胞代谢需要并降低血浆胆固醇含量(图)。人LDL受体为160kD的糖蛋白,由839个氨基酸残基组成,其编码基因位于19号染色体上。FH是由于基因突变引起的LDL受体缺陷症,为常染色体显性遗传,按LDL受体突变的类型及分子机制可分为:
目前已发现LDL受体有150多种突变,包括基因缺失与插入、错义与无义突变等,可干扰受体代谢的各个环节。按LDL受体突变的类型及分子机制可分为:
①受体合成受损(impairment of receptor synthesis)。由于上游外显子及内含子的大片缺失使受体转录障碍;基因重排造成阅读框架移位,使编码氨基酸的密码子变成终止密码等,使之不能编码正常的受体蛋白;
②细胞内转运障碍(impairment of the receptor movement)。受体前体滞留在高尔基体,不能转变为成熟的受体以及向细胞膜转运受阻,受体在内质网内被降解;
③受体与配体结合力降低(reduced capacity of receptor to bind lipoprotein)。由于编码配体结合区的碱基缺失或突变,细胞膜表面的LDL受体不能与LDL结合或结合力降低;
④受体内吞缺陷(impairment of receptor clustering and internalization)。因编码受体胞浆区的基因突变,与LDL结合的受体不能聚集成簇,或不能携带LDL进入细胞;
⑤受体再循环障碍(impairment of receptor recycling)。基因突变使内吞的受体不能在酸性pH下与LDL解离,受体在细胞内降解,不能参与再循环。
因LDL受体数量减少或功能异常,其对血浆LDL的清除能力降低,患者出生后血浆LDL含量即高于正常,发生动脉粥样硬化的危险也显著升高。纯合子FH系编码LDL受体的等位基因均有缺陷,发病率约1/100万,患者LDL受体缺失或严重不足,血浆LDL水平可高达正常人的6倍,并有早发的动脉粥样硬化,在儿童期即可出现冠状动脉狭窄和心绞痛,常在20岁前就因严重的动脉粥样硬化而过早死亡;杂合子FH为编码LDL受体等位基因的单个基因突变所致,发病率约为1/500,患者的LDL受体量为正常人的一半,血浆LDL含量约为正常人的2~3倍,患者多于40~50岁发生冠心病。
2.家族性肾性尿崩症
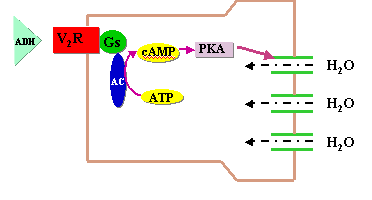
因肾小管对ADH 反应性降低引起的尿崩症称为肾性尿崩症,可由遗传性ADH受体及受体后信息传递异常或继发性肾及肾外疾病引起,前者称为家族性肾性尿崩症。ADH V2受体位于远端肾小管或集合管上皮细胞膜。当ADH与受体结合后,经激活Gs增加AC活性,在PKA的催化下,使微丝微管磷酸化,促进位于胞浆内的水通道蛋白向集合管上皮细胞管腔侧膜移动并插入膜内,集合管上皮细胞膜对水的通透性增加,管腔内水进入细胞,并按渗透梯度转移到肾间质,使肾小管腔内尿液浓缩(图)。
编码人ADH受体的基因位于X染色体,故家族性肾性尿崩症系性连锁遗传,其发病机制是由于基因突变使ADH受体合成减少或受体胞外环结构异常。无论是受体数量不足或亲和力降低,均使ADH对远端肾小管和集合管上皮细胞的刺激作用减弱,cAMP生成减少,对水的重吸收降低。家族性肾性尿崩症患者多在1岁以内发病,男性显示症状,具有口渴、多饮、多尿等尿崩症的临床特征,但血中ADH水平在正常水平以上,女性携带者一般无症状。
部分家族性肾性尿崩症患者可出现ADH受体后调节异常,此为常染色体隐性遗传,因水通道蛋白缺陷使肾小管对ADH敏感性降低,但在临床上与ADH受体缺陷很难区分。
3.甲状腺素抵抗综合征
因靶细胞对激素反应性降低或丧失而引起的一系列病理变化称为激素抵抗综合征(hormone resistance syndrome),临床表现以相应激素的作用减弱为特征,但循环血中该激素水平升高。人类有α和β两型甲状腺素受体,分别由独立基因编码。目前已发现编码β型受体的基因突变使外周组织对甲状腺素抵抗,但迄今尚未发现α型受体基因突变与甲状腺素抵抗综合征有关。有缺陷的甲状腺素受体不能与T3结合,难以调节含甲状腺素反应元件的基因转录。患者的临床表现取决于突变受体的数量,可从轻微的甲状腺素不足到严重的甲状腺功能减退,甚至影响生长发育,血中T3和T4水平升高。
(二)自身免疫性受体病
因体内产生抗受体的自身抗体而引起的疾病,可因刺激性抗体引起细胞对配体的反应性增强,或因阻断性抗体干扰配体与受体的结合,导致细胞的反应性降低。
1.重症肌无力
重症肌无力是一种神经肌肉间传递功能障碍的自身免疫病,主要特征为受累横纹肌稍行活动后即迅速疲乏无力,经休息后肌力有程度不同的恢复。轻者仅累及眼肌,重者可波及全身肌肉,甚至因呼吸肌受累而危及生命。
正常情况下,当神经冲动抵达运动神经末梢时,后者释放乙酰胆碱(Ach),Ach与骨骼肌的运动终板膜表面的烟碱型乙酰胆碱(n-Ach)受体结合,使受体构型改变,离子通道开放,Na+内流形成动作电位,肌纤维收缩。在患者的胸腺上皮细胞及淋巴细胞内含有一种与n-Ach受体结构相似的物质,其可能作为自身抗原而引起胸腺产生抗n-Ach受体的抗体。在实验性重症肌无力动物或临床重症肌无力患者的血清中可检测到抗n-Ach受体的抗体,其含量与疾病的严重程度呈平行关系。将重症肌无力患者的血浆注射给小鼠,可诱发类似重症肌无力的变化。抗 n-Ach受体抗体通过干扰Ach与受体的结合;或是加速受体的内吞与破坏,最终导致运动神经末梢释放的Ach不能充分与运动终板上的n-Ach受体结合,使兴奋从神经传递到肌肉的过程发生障碍,从而影响肌肉的收缩。
2.自身免疫性甲状腺病
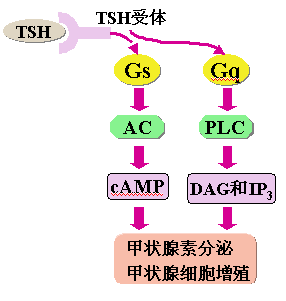
促甲状腺素(TSH)是腺垂体合成和释放的糖蛋白激素,它与甲状腺细胞膜上的TSH受体相结合,经Gs激活AC,增加cAMP生成;亦可经Gq介导的PLC增加DG和IP3生成,其生物学效应是调节甲状腺细胞生长和甲状腺素分泌(图)。
多种甲状腺自身抗体均可引起自身免疫性甲状腺病,根据自身抗体的性质不同,患者的临床表现各异。TSH受体抗体分为两种:①刺激性抗体 其与TSH 受体结合后能模拟TSH 的作用,通过激活G蛋白,促进甲状腺素分泌和甲状腺腺体生长,在Graves病(弥漫性甲状腺肿)患者血中可检出TSH受体刺激性抗体,与甲状腺功能亢进和甲状腺肿大的临床表现有关;②阻断性抗体 阻断性抗体可存在于(慢性淋巴细胞性甲状腺炎)和特发性粘液性水肿患者血中,其与TSH受体的结合减少了TSH与受体的结合,减弱或消除了TSH的作用,抑制甲状腺素分泌,造成甲状腺功能减退。近年来的研究表明,刺激性和阻断性抗体都可与TSH受体的胞外区结合,但刺激性抗体与TSH受体30~35位氨基酸残基结合,而阻断性抗体则与受体295~302和385~395位氨基酸残基结合,这种与TSH受体结合部位的不同为解释Graves病和桥本病临床特征的差异提供了分子基础。
(三)继发性受体异常
许多疾病过程中,可因配体的含量、pH、磷脂膜环境及细胞合成与分解蛋白质的能力等变化引起受体数量及亲和力的继发性改变。其中有的是损伤性变化,如膜磷脂分解引起受体功能降低;有的是代偿性调节,如配体含量增高引起的受体减敏,以减轻配体对细胞的过度刺激。继发性受体异常又可进一步影响疾病的进程。
例如,肾上腺素能受体及其细胞内信号转导是介导正常及心力衰竭时心功能调控的重要途径。正常人心肌细胞膜含β1、β2和α1肾上腺素能受体,其中β1受体占70%~80%,是调节正常心功能的主要肾上腺素能受体亚型。已有大量研究表明,心力衰竭患者及动物的心脏对异丙肾上腺素引起的正性肌力反应明显减弱,即β受体对儿茶酚胺刺激发生了减敏反应。心力衰竭时,β受体下调,特别是β1受体数量减少,可降至50%以下;β2受体数量变化不明显,但对配体的敏感性亦有降低。β受体减敏是对过量儿茶酚胺刺激的代偿反应,可抑制心肌收缩力,减轻心肌的损伤,但也是促进心力衰竭发展的原因之一。此外,受体后信号转导异常,如Gi/Gs比例升高,亦在心功能障碍中起作用。
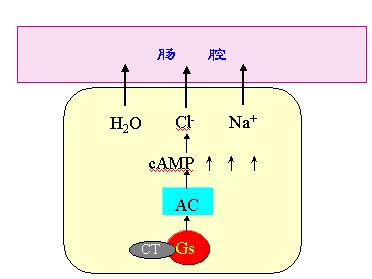
(一) 霍乱(Cholera)
霍乱是由霍乱弧菌引起的烈性肠道传染病。患者起病急骤,剧烈腹泻,常有严重脱水、电解质紊乱和酸中毒,可因循环衰竭而死亡。霍乱弧菌通过分泌活性极强的外毒素-霍乱毒素干扰细胞内信号转导过程。霍乱毒素选择性催化Gsα亚基的精氨酸201核糖化,此时Gsα仍可与GTP结合,但GTP酶活性丧失,不能将GTP水解成GDP,从而使Gsα处于不可逆性激活状态,不断刺激AC生成cAMP,胞浆中的cAMP含量可增加至正常的100倍以上,导致小肠上皮细胞膜蛋白构型改变,大量氯离子和水分子持续转运入肠腔,引起严重的腹泻和脱水(图)。
(二)假性甲状旁腺功能减退症(pseudohypoparathyroidism, PHP)
PHP是由于靶器官对甲状旁腺激素(PTH)反应性降低而引起的遗传性疾病。PTH受体与Gs偶联,经激活AC催化cAMP生成,其作用为:①促进远端肾小管重吸收钙;②抑制近端肾小管重吸收磷酸盐;③促进肾小管产生1,25(OH)2D3,后者作用于肠粘膜细胞,增加钙的吸收;④促进骨钙和骨磷酸盐释放,维持细胞外液钙浓度。
PHP可分为两型:①PHP1A型 其发病机制是由于编码Gsα等位基因的单个基因突变,患者Gsα mRNA可比正常人降低50%,导致PTH受体与AC之间信号转导脱偶联。Gs基因突变还可引起其他与AC相连的激素抵抗症,如TSH、LH和FSH抵抗症等,部分患者还伴有奥尔布赖特(Albright) 遗传性骨营养不良的表现,如身材矮小、短颈、圆脸和短指畸形等(图);②PHP1B型 此型患者仅对PTH抵抗,Gs正常,也与PTH受体的遗传学无关,具体发病机制不明。PHP患者由于肾小管对磷重吸收增加,血磷升高;肾小管对钙重吸收减少及1,25(OH)2D3生成减少,尿钙升高和血钙降低,患者血浆PTH继发性升高,但靶器官对PTH 无反应。
(三)肢端肥大症(acromegaly)和巨人症(gigantism)
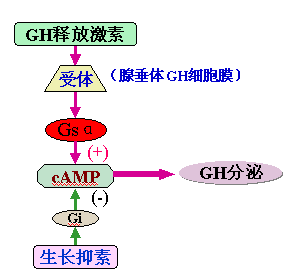
生长激素(growth hormone,GH)是腺垂体分泌的多肽激素,其功能是促进机体生长。GH的分泌受下丘脑GH释放激素和生长抑素的调节,GH释放激素经激活Gs,导致AC活性升高和cAMP积聚,cAMP可促进分泌GH的细胞增殖和分泌(图);生长抑素则通过减少cAMP水平抑制GH分泌。分泌GH过多的垂体腺瘤中,有30%~40%是由于编码Gsα的基因点突变,其特征是Gsα的精氨酸201为半胱氨酸或组氨酸所取代,或谷氨酰胺227为精氨酸或亮氨酸所取代,这些突变抑制了GTP酶活性,使Gsα处于持续激活状态,AC活性升高,cAMP含量增加,垂体细胞生长和分泌功能活跃。故在这些垂体腺瘤中,信号转导障碍的关键环节是Gsα过度激活导致的GH释放激素和生长抑素对GH分泌的调节失衡。GH的过度分泌,可刺激骨骼过度生长,在成人引起肢端肥大症,在儿童引起巨人症。
在某些疾病时,Gs或Gi还可发生继发性变化,影响细胞信号转导过程。
四、多个环节细胞信号转导障碍与疾病
在许多疾病过程中,细胞信号转导异常不仅可发生在某一信息分子或单一信号转导途径,亦可先后或同时涉及多个信息分子并影响多个信号转导过程,导致复杂的网络调节失衡,促进疾病的发生与发展。
(一)非胰岛素依赖性糖尿病
非胰岛素依赖性糖尿病(non-insulin dependent diabetes mellitus,NIDDM)又称II型糖尿病,患者除血糖升高外,血中胰岛素含量可增高、正常或轻度降低,80%患者伴有肥胖。胰岛素受体前、受体和受体后异常是造成细胞对胰岛素反应性降低的主要原因,其中与信号转导障碍有关的是:
1.胰岛素受体异常
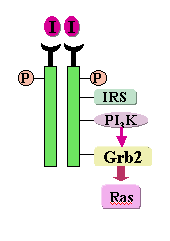
胰岛素受体属于受体TPK家族,由α、β亚单位组成。与PDGF受体不同,无活性的胰岛素受体在未与配体结合时即以二聚体的形式存在于细胞膜。胰岛素与受体α亚单位结合后,引起β亚单位的酪氨酸磷酸化,并在胰岛素受体底物1/2(insulin receptor substrate 1/2, IRS-1/2)的参与下,与含SH2区的Grb2和PI3K结合,启动与代谢和生长有关的下游信号转导过程(图)。根据胰岛素受体异常的原因可分为:
(1)遗传性胰岛素受体异常:因基因突变所致,包括:①受体合成减少或结构异常的受体在细胞内分解破坏增多导致受体数量减少;②受体与配体的亲和力降低,如受体精氨酸735突变为丝氨酸,使合成的受体不能正确折叠,与胰岛素亲和力下降;③受体TPK活性降低,如甘氨酸1008突变为缬氨酸,胞内区TPK结构异常,磷酸化酪氨酸的能力减弱。
(2)自身免疫性胰岛素受体异常:血液中存在抗胰岛素受体的抗体。
(3)继发性胰岛素受体异常:任何原因引起的高胰岛素血症均可使胰岛素受体继发性下调,引起胰岛素抵抗综合征。
2.受体后信号转导异常
目前认为PI3K做为一个传递受体TPK活性到调节丝/苏氨酸蛋白激酶的级联反应的分子开关,在胰岛素上游信号转导中具有重要作用。在非胰岛素依赖性ob/ob小鼠糖尿病模型中,可见胰岛素对PI3K活性的刺激作用明显降低,肝细胞p85含量降低50%;而在胰岛素依赖性糖尿病鼠则未见PI3K的抑制。II型糖尿病患者的肌肉和脂肪组织也可见胰岛素对PI3K的激活作用减弱。PI3K基因突变可产生胰岛素抵抗,目前已发现在p85基因有突变,但尚未发现p110的改变。胰岛素受体后信号转导异常除因PI3K表达的改变外,也与IRS-1和IRS-2的下调使胰岛素引起的经PI3K介导的信号转导过程受阻有关,在敲除IRS-2基因的小鼠可见胰岛素对肌肉和肝细胞PI3K的刺激作用降低,提示受体后信号转导障碍可发生在IRS和PI3K两个环节。
(二) 高血压病
正常血管平滑肌细胞(vascular smooth muscle cell, VSMC)呈非增殖性的收缩表型,在神经及激素刺激下调节血管壁张力,维持组织血流量。在病理状态下,VSMC转化为合成表型,生成和分泌多种血管活性物质、生长因子及细胞外基质,同时自身发生迁移、肥大(hypertrophy,指细胞体积增大)和增殖(hyperplasia,指细胞数量增加)。VSMC生物学变化可引起血管壁增厚、管腔狭窄、血管顺应性降低和血管重构,在高血压病的发生与发展中起重要作用。引起VSMC增殖肥大的细胞外信息可分为:
1.生物化学性因素对VSMC的刺激
指由内分泌、旁分泌或自分泌作用于VSMC的激素、细胞因子及生长因子等。在许多心血管疾病患者或动物模型中,可以检测到血浆或血管壁局部血管活性物质和促生长因子的含量升高,它们除改变血管口径外,还有很强的促肥大增殖作用。去甲肾上腺素、血管紧张素II和内皮素分别与各自的受体结合后,激活Gq及PLCβ,经磷脂酰肌醇级联反应,改变细胞的功能与代谢并增强基因转录。生长因子如PDGF等通过作用于细胞表面的受体TPK,引起受体酪氨酸残基自身磷酸化,经与一系列信号转导分子的相互作用,引起基因转录和蛋白质合成增加。
2.机械性因素对血管壁细胞的刺激
血液在血管内流动和血管内压的周期性变化对内皮细胞和VSMC产生机械性刺激,这对维持血管稳态起重要作用,同时也是导致VSMC增殖肥大的病理生理学因素。一般而言,机械力的急性变化通过释放血管活性物质调节血管口径;而长期的机械性刺激则通过促进细胞增殖肥大,引起血管壁增厚和血管重构。
整合素(integrin)是一组介导细胞与细胞间及细胞与细胞外基质之间粘附反应的跨膜糖蛋白,机械性刺激可促进细胞外基质蛋白如纤维粘连蛋白、胶原蛋白与血管壁细胞表面的整合素相连,再经与细胞内骨架蛋白的相互作用,形成以整合素为中心的粘附点,进而激活粘附点激酶(focal adhesion kinase,FAK)。已有实验报道在机械力刺激下,血管内皮细胞和VSMC的整合素表达增强;抑制细胞外基质与整合素结合可消除机械力引起的血管内皮细胞酪氨酸磷酸化;应用整合素抗体亦可消除血管壁细胞的磷酸化反应,这些都说明整合素在血管壁细胞感受机械性刺激中起重要作用。FAK是125kD的酪氨酸激酶,其下游信号分子尚未完全明确。有实验表明FAK磷酸化后暴露出与Grb2相结合的位点,经形成Grb2-Sos复合物激活下游的ERK。
实验表明机械性和生物化学性刺激均可引起血管壁细胞中Ras含量迅速升高,无活性突变的Ras可减弱机械力刺激引起的ERK激活,急性高血压和球囊损伤血管壁亦可激活VSMC内ERK。激活的ERK 转移入核,磷酸化转录因子,进而影响调节促增殖肥大和细胞周期的基因表达。在ERK激活的同时,可见AP-1转录因子激活和c-fos、c-jun表达增加。FAK还可和PLCγ亚型相连并使之激活,通过促进膜磷脂分解而影响细胞生物学活性。
在病理状态下,各种促肥大增殖因素不但可独立发挥作用,而且还可相互影响。例如,血管活性物质和生长因子作用于VSMC,可促进其生成促增殖肥大因子,并增强血管内皮细胞和VSMC对机械性刺激的敏感性;而受到机械力作用的血管壁细胞又可生成和释放血管活性物质和生长因子,导致细胞内多种信号转导分子的激活及各信号转导途径之间的交互调节,形成复杂的网络联系,共同引起细胞肥大与增殖。
(三)细胞信号转导障碍与肿瘤
正常细胞的生长与分化受到精细的网络调节,细胞癌变最基本的特征是生长失控及分化异常。近年来人们认识到绝大多数的癌基因表达产物都是细胞信号转导系统的组成成分,它们可以从多个环节干扰细胞信号转导过程,导致肿瘤细胞增殖与分化异常。
1.表达生长因子样物质
某些癌基因可以编码生长因子样的活性物质,例如,sis癌基因的表达产物与PDGFβ链高度同源,int-2癌基因蛋白与成纤维细胞生长因子结构相似。此类癌基因激活可使生长因子样物质生成增多,以自分泌或旁分泌方式刺激细胞增殖。在人神经胶质母细胞瘤、骨肉瘤和纤维肉瘤中均可见sis基因异常表达。
2.表达生长因子受体类蛋白
某些癌基因可以表达生长因子受体的类似物,通过模拟生长因子的功能受体起到促增殖的作用。例如,erb-B癌基因编码的变异型EGF受体,缺乏与配体结合的膜外区,但在没有EGF存在的条件下,就可持续激活下游的增殖信号。在人乳腺癌、肺癌、胰腺癌和卵巢肿瘤中已发现EGF受体的过度表达;在卵巢肿瘤亦可见PDGF受体高表达,且这些受体的表达与预后呈负相关。
3.表达蛋白激酶类
某些癌基因可通过编码非受体TPK或丝/苏氨酸激酶类影响细胞信号转导过程。例如,src癌基因产物具有较高的TPK活性,在某些肿瘤中其表达增加,可催化下游信号转导分子的酪氨酸磷酸化,促进细胞异常增殖。此外,还使糖酵解酶磷酸化,糖酵解酶活性增加,糖酵解增强是肿瘤细胞的代谢特点之一。mos、raf癌基因编码丝/苏氨酸蛋白激酶类产物,其可促进MAPK磷酸化,进而促进核内癌基因表达。
4.表达信号转导分子类
ras癌基因编码的21kD小分子G蛋白Ras,可在Sos催化下通过与GTP结合而激活下游信号转导分子。在30%的人肿瘤组织已发现有不同性质的ras基因突变,其中突变率较高的是甘氨酸12、甘氨酸13或谷氨酰胺61为其他氨基酸残基所取代。变异的Ras与GDP解离速率增加或GTP酶活性降低,均可导致Ras持续活化,促增殖信号增强而发生肿瘤。例如,人膀胱癌细胞ras基因编码序列第35位核苷酸由正常G突变为C,相应的Ras蛋白甘氨酸12突变为缬氨酸,使其处于持续激活状态。
5.表达核内蛋白类
某些癌基因如myc、fos、jun的表达产物位于核内,能与DNA结合,具有直接调节转录活性的转录因子样作用。过度表达的癌基因可引起肿瘤发生,如高表达的jun蛋白与fos蛋白与DNA上的AP-1位点结合,激活基因转录,促进肿瘤发生。
综上所述,细胞信号转导障碍对疾病的发生发展具有多方面的影响,其发生原因是多种多样的,基因突变、细菌毒素、细胞因子、自身抗体和应激等均可以造成细胞信号转导过程的原发性损伤,或可引起它们的继发性改变。细胞信号转导障碍可以局限于单一环节,亦可同时或先后累及多个环节甚至多条信号转导途径,造成调节信号转导的网络失衡,引起复杂多变的表现形式。细胞信号转导障碍在疾病中的作用亦表现为多样性,既可以做为疾病的直接原因,引起特定疾病的发生;亦可干扰疾病的某个环节,导致特异性症状或体征的产生。细胞信号转导障碍还可介导某些非特异性反应,出现在不同的疾病过程中。随着研究的不断深入,已经发现越来越多的疾病或病理过程中存在着信号转导异常,认识其变化规律及其在疾病发生发展中的病理生理意义,不但可以揭示疾病的分子机制,而且为疾病的防治提出了新的方向。
细胞增殖异常和细胞周期调控障碍的分子机制不但涉及肿瘤的发生,而且与动脉粥样硬化、血管成型术后再狭窄等增殖性疾病(proliferative diseases)的发生有关,亦在炎症反应如脓毒血症、类风湿性关节炎及组织排斥反应中起作用。因而,有学者采用信号转导治疗(signal transduction therapy)的方法,以信号转导蛋白为靶分子对疾病进行防治。
目前研究较集中的是抑制酪氨酸蛋白激酶介导的细胞信号转导途径。由于85%与肿瘤相关的原癌基因和癌基因产物是TPK,且肿瘤时TPK活性常常升高,故以TPK为靶分子可阻断细胞增殖。①采用单克隆抗体阻断配体与受体TPK结合:实验表明,抗EGF受体胞外区的单克隆抗体能有效抑制人鳞状细胞癌在裸鼠体内的生长和转移,并延长裸鼠生存期;目前,抗EGF受体的单克隆抗体已用于肿瘤的临床实验治疗;②抑制TPK的催化活性: 信号转导的研究为药物干预开创了一个新领域,设计特异性抑制TPK活性和细胞生长的药物,不但是开发抗肿瘤药物的重要方向,而且在球囊引起的血管损伤的大鼠或猪,局部应用选择性或非选择性TPK抑制剂,能有效阻止血管平滑肌增殖和迁移,减轻或防止再狭窄的发生。
Ras是介导TPK信号转导途径的关键分子,抑制Ras向膜转移可阻断其激活,或应用无活性突变的Ras阻断Ras信号转导过程,是正在探索的肿瘤治疗方法。此外,还有一些针对细胞周期调控、转录因子和核受体环节干扰信号转导途径的措施正在研究中。