
肾功能不全(renal insufficiency)是指各种原因引起肾脏泌尿功能严重障碍,使代谢产物及毒性物质不能排出体外,以致产生水、电解质和酸碱平衡紊乱,并伴有肾脏内分泌功能障碍的综合征。
肾功能不全与肾功能衰竭(renal failure)的本质相同,只是程度上有区别。肾功能不全包括肾功能障碍从轻到重的全过程,而肾功能衰竭则是肾功能不全的晚期阶段。但在临床应用中,二者往往通用。
急性肾功能不全(acute renal insufficiency,ARI)是指各种原因引起肾脏泌尿功能在短期内急剧降低,以致不能维持内环境稳定,从而引起水、电解质和酸碱平衡紊乱以及代谢产物蓄积的综合征。
通常将急性肾功能不全的病因分为三类:肾前性、肾性和肾后性。
| 急性肾功能不全分类 | |
| 类别 | 原因 |
| 肾前性 | 肾血液灌注减少 |
| 肾性 | 肾实质病变,如急性肾小管坏死 |
| 肾后性 | 肾以下尿路阻塞 |
(一)肾前性急性肾功能不全(Prerenal acute renal insufficiency)
任何原因引起的肾血液灌流量急剧减少而导致的泌尿功能障碍,此时肾无器质性病变,如肾灌流量及时恢复,则肾功能也随即恢复正常,又称为功能性急性肾功能不全(functional acute renal insufficiency)。
| 肾前性急性肾功能不全的病因 | ||||||
|
(二)肾性急性肾功能不全(Intrarenal acute renal insufficiency)
由于各种原因引起肾实质病变而产生的急性肾功能不全,又称器质性急性肾功能不全(parenchymal acute renal insufficiency)。
| 肾性急性肾功能不全的病因 | ||||
|
(三)肾后性急性肾功能不全(Postrenal acute renal insufficiency)
由于肾以下尿路(从肾盏到尿道口任何部位)梗阻引起的急性肾功能不全,称为肾后性急性肾功能不全(Postrenal acute renal insufficiency)。
| 肾后性急性肾功能不全的病因 | ||
|
二、发病机制(Pathogenesis)
各种原因引起的急性肾功能不全的发病机制有所不同,且其机制尚未彻底阐明。下面主要阐述肾缺血和肾毒物引起少尿型急性肾功能不全的发病机制。
(一)肾缺血(Renal ischemia)
目前一般认为,持续性肾缺血和肾血流量远离皮质分布是ARI初期的主要发病机制。造成肾缺血主要与肾灌注压降低、肾血管收缩和肾血液流变学变化有关。
1.肾灌注压下降(Decrease in renal perfusion pressure)
肾血流量有自身调节,当动脉血压在80~180mmHg范围内变动时,肾血管通过自身调节,使肾血流量和肾小球滤过率(glomerular filtration rate, GFR)保持稳定。肾前性ARI时,动脉血压常低于80mmHg,肾血管失去自身调节,肾血流量减少,肾小球毛细血管血压下降,使肾小球有效滤过压减小,导致少尿或无尿。
2.肾血管收缩(Contraction of renal blood vessels)
(1)肾素-血管紧张素系统活性增高
| 1、肾缺血或肾中毒时,近曲小管上皮细胞受损,对Na+重吸收减少,到达远曲小管尿液中的Na+浓度升高,刺激致近球细胞分泌肾素; |
| 2、缺血时肾灌注压降低,入球小动脉管壁张力下降,刺激近球细胞分泌肾素; |
| 3、有效循环血量降低,交感神经兴奋的直接刺激等均可引起肾素分泌增加,继而血管紧张素Ⅱ增加,使肾血管收缩,从而导致GFR降低。 |
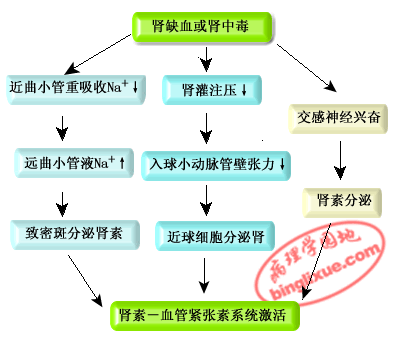 |
(2)体内儿茶酚胺增加
休克或创伤引起的急性肾功能不全时,机体交感-肾上腺髓质系统兴奋,血中儿茶酚胺急剧增加。皮质肾单位的入球小动脉对儿茶酚胺敏感性高,故急性肾功能不全时以肾皮质外层血流量减少最为明显,称为肾内血流分布异常。
(3)肾髓质间质细胞合成前列腺素减少
肾缺血、肾中毒时,肾髓质间质细胞合成前列腺素减少,PGE2合成减少,TXA2相对增加,结果导致肾血管痉挛、收缩,阻力增加并且微血管内血栓形成和阻塞。
3.血流变学的变化(Alteration in hemorheology)
(1)血液粘滞度升高
ARI时,血中纤维蛋白原增多、红细胞聚集及其变形能力下降、红细胞破裂及血红蛋白释出、血小板聚集等均可引起血液粘滞度升高,影响肾小球毛细血管床的微循环状态,造成肾小球滤过率下降。
(2)白细胞与肾血管阻力
ARI时,由于白细胞变形能力降低,粘附血管壁能力增高,造成微血管阻塞,血流阻力增加,微循环灌流量减少。
(3)微血管改变
ARI时,肾微血管口径缩小、自动调节功能丧失、血红蛋白附壁,这些变化使肾微血管痉挛、增厚,加重肾缺血。
(二)肾小管阻塞(Obstruction of renal tubules)
肾缺血、肾毒物引起急性肾小管坏死后脱落的细胞及碎片可阻塞肾小管,溶血性疾患或挤压综合征使大量血红蛋白、肌红蛋白在肾小管内形成管型,其它如多发性骨髓瘤本周蛋白、磺胺等药物结晶,均可沉积在肾小管管腔内,造成广泛的肾小管阻塞,使原尿不易通过,形成少尿;同时,由于管腔内压升高,使有效滤过压降低,导致肾小球滤过率降低。管型阻塞是ARI持续期导致GFR减少的重要因素。
(三)肾小管原尿返流(Reflux of urine)
ATN时,肾小管上皮细胞广泛坏死,基底膜断裂,原尿经断裂的基底膜扩散到肾间质,直接造成尿量减少,而且回漏的原尿能引起肾间质水肿,压迫肾小管和肾小管周围的毛细血管。肾小管受压,阻塞加重,阻碍原尿在肾小管内通过并造成囊内压升高,使肾小球有效滤过压进一步降低;毛细血管受压,使肾小管供血进一步减少,导致肾损伤加重,形成恶性循环。
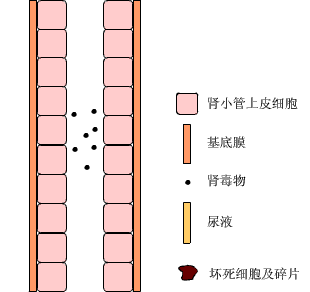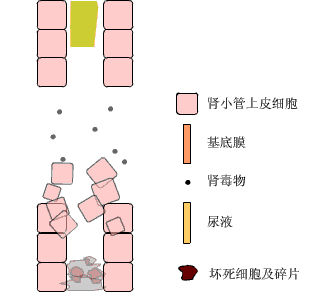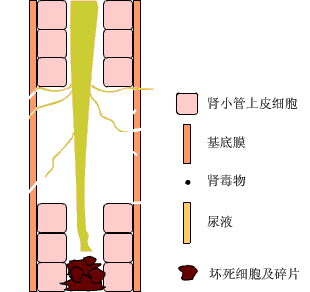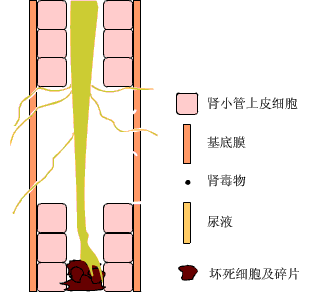 |
(四) 肾组织细胞损伤及机制(Injury of nephric cell)
肾内各种细胞(包括肾小管细胞、内皮细胞、系膜细胞等)受损而出现的代谢、功能及形态结构的紊乱是ARI时GFR持续降低、内环境紊乱的基本机制。
1.受损细胞(injured cell)
(1)肾小管细胞 肾小管细胞损伤在ARI的发生发展中起重要作用。
ARI有两种肾小管损伤的病理特征,即小管破裂性损伤(tubulorrhexic lesion)和肾毒性损伤(nephrotoxic lesion)。小管破裂性损伤表现为肾小管上皮细胞坏死、脱落,基底膜被破坏,肾小管各段都可受累,但并非每个肾单位都出现损伤,肾中毒及肾持续缺血的病例均可见到这种损伤。肾毒性损伤表现为肾小管上皮细胞大片坏死,但基底膜完整,损伤部位主要为近球小管,可累及所有肾单位,见于肾中毒的病例。
(2)内皮细胞 肾血管内皮细胞肿胀、功能受损均促进ARI的发生与发展。
内皮细胞肿胀,血管管腔变窄,血流阻力增加,肾血流减少;内皮细胞受损激发血小板聚集与微血栓形成以及毛细血管内凝血;肾小球内皮细胞窗变小甚至减少可直接影响超滤系数;内皮细胞释放舒血管因子减少,缩血管因子增多均可加强肾血管的持续收缩。以上因素均可导致GFR持续降低。
2.细胞损伤的机制(mechanism of cell injury)
ARI时,肾小管细胞可因缺血、毒物或二者的共同作用引起损伤。细胞内ATP储备不足是缺血性损伤的主要因素。细胞ATP不足引起依赖ATP的膜转运系统功能变化,导致细胞内游离钙增高、活性氧的损伤及磷脂酶的异常激活等。与细胞损伤机制相关的因素有:
| 1、ATP产生减少及Na+、K+-ATP酶活性降低。缺血时因缺氧、代谢底物缺乏、线粒体氧化磷酸化速度减慢,导致ATP生成障碍。ATP减少不仅减弱肾小管的主动重吸收功能,而且由于Na+、K+-ATP酶活性减弱,细胞内Na+、水潴留,导致细胞水肿;由于Ca2+-ATP酶活性减弱及Na+-Ca2+交换的增强,造成细胞内Ca2+超载。细胞内游离钙增加又加重线粒体的损伤,使ATP生成更加减少,从而形成恶性循环导致细胞死亡。 |
| 2、自由基产生增多与清除减少。肾缺血及缺血后再灌注均可使自由基产生增加;由于缺血引起内源性自由基清除系统如过氧化物酶缺乏而使自由基清除减少。自由基过多导致脂质过氧化、细胞蛋白的氧化及DNA的损伤,造成质膜完整性的破坏、蛋白功能丧失及细胞损伤修复的障碍。 |
| 3、磷脂酶活性增高。缺血性损伤过程中,细胞内Ca2+浓度升高,还原型谷胱甘肽(GSH)减少,导致磷脂酶A2活性增高,细胞膜被降解,细胞骨架结构解体。 |
 |
三、临床表现及病理生理学基础(Clinical manifestations and pathophysilogical base)
急性肾功能不全在临床上表现为两种类型,即少尿型和多尿型。
(一)少尿型急性肾功能不全(Oliguric ARI)
少尿型急性肾功能衰竭的发生发展可分为三个阶段,即少尿期、多尿期和恢复期。
1.少尿期(oliguric phase)
(1)尿的变化
尿量:多数ARI患者尿量迅速减少,通常表现为少尿(24h尿量少于400ml)或无尿(24h尿量少于100ml)。 临床上,虽然功能性ARI和器质性ARI都表现出少尿,但两者在少尿发生机制及尿液成分上均有区别。鉴别ARI是功能性还是器质性,对于指导临床治疗和估计预后都有重要意义。
|
功能性急性肾功能不全 | 器质性急性肾功能不全 | 机制 |
| 尿比重 | >1.020 | <1.015 | 功能性ARI时,肾小管对水的重吸收增加;器质性ARI时,肾小管对水的重吸收功能降低 |
| 尿渗透压(mmol/L) | >700 | <250 | |
| 尿钠含量(mmol/L) | <20 | >40 | 器质性ARI时,肾小管上皮细胞受损,对原尿中Na+重吸收障碍 |
| 尿肌酐/血肌酐 | >40 | <10 | |
| 尿蛋白及镜检 | 正常 | 尿蛋白(+)、各种管型、红细胞、上皮细胞 | ARI时肾小球滤过功能障碍和肾小管上皮坏死脱落 |
| 补液实验 | 尿量增加 | 尿量不增加 | |
| 输液原则 | 充分扩容 | 量出为入 |
(2)水中毒
ARI患者发生水中毒的原因:少尿或无尿;机体分解代谢增强、内生水增加;摄入或输入液体过多。
水中毒使细胞外液呈低渗状态,水分向细胞内转移引起细胞内水肿,严重时患者可出现心功能不全、脑水肿和肺水肿。
(3)高钾血症
高钾血症是ARI患者在少尿期最危险的并发症,是少尿期一周内病人死亡的主要原因。高钾血症可引起传导阻滞和诱发心律失常,严重时出现心室颤动或心脏骤停。
原因 |
结果 |
少尿、无尿 |
排钾减少 |
使用保钾利尿剂 |
|
摄入含钾过多的药物、食物 |
摄钾增多 |
输入库存血 |
|
组织损伤和分解代谢增强 |
钾从细胞内释出 |
(4)代谢性酸中毒
| ARI患者代谢性酸中毒的发生机制 | |
原因 |
结果 |
肾小球滤过率降低 |
酸性产物排出减少 |
肾小管泌H+及泌NH3能力降低 |
NaHCO3重吸收障碍 |
分解代谢增强 |
固定酸产生增加 |
(5)氮质血症
肾功能不全时,由于肾小球滤过率下降,含氮的代谢终产物如尿素、肌酐、尿酸等在体内堆积,使血中非蛋白质氮(nonprotein nitrogen,NPN)的含量显著升高(>28.6mmol/L,相当于>40mg/dl) ,称为氮质血症(azotemia)。
正常人血中有九种非蛋白含氮化合物,其中尿素、尿酸和肌酐必须通过肾排出体外。当肾功能不全时,这三种化合物特别是尿素和肌酐在血中浓度升高,故临床上常用血尿素氮和血肌酐浓度作为氮质血症的指标。ARI少尿期一开始,血中NPN即明显增高,如合并感染、中毒、烧伤、创伤或摄入过多高蛋白饮食时,可加重氮质血症。
2.多尿期(diuretic phase)
ARI患者每天尿量超过400ml即进入多尿期,说明病情趋向好转,此期可持续2周。产生多尿的机制是:
| ①肾血流量和肾小球滤过功能逐渐恢复; |
| ②肾小管上皮细胞虽已开始再生修复,但其重吸收功能尚不完善,原尿不能充分浓缩; |
| ③肾间质水肿消退,肾小管阻塞解除; |
| ④少尿期滞留的尿素经肾小球滤过增多,引起渗透性利尿; |
| ⑤少尿期大量水分在体内潴留,当肾功能逐渐恢复时,肾代偿性排出体内多余水分。 |
此期由于水、电解质大量排出,如不即时补充,则可发生脱水、低钾血症和低钠血症,因此,在多尿期仍需控制和调整摄入的水和电解质的量。
3.恢复期(recovery phase)
一般在发病后一个月进入恢复期,肾功能恢复正常约需要3个月到1年时间。此期患者的尿量基本恢复正常,代谢产物的潴留和水、电解质、酸碱平衡紊乱得到纠正,但肾小管浓缩功能完全恢复正常需要较长时间。少数患者由于肾小管上皮细胞破坏严重和修复不全,可能转变为慢性肾功能不全。
(二)非少尿型急性肾功能不全(Non-oliguric ARI)
非少尿型ARI患者临床表现一般较轻,病程较短,并发症少,病死率低,预后较好。其主要特点是:
| ① 无明显少尿 |
| ② 尿比重低,尿钠含量低 |
| ③ 有氮质血症 |
| ④ 多无高钾血症 |
机制:非少尿型ARI的病理损害较轻,肾小球滤过率下降不如少尿型严重;肾小管损伤也较轻,主要表现为尿浓缩功能障碍,所以尿量相对较多。
四、急性肾功能不全的防治原则(Treatment principles)
(一)预防(Prophylaxis)
1.合理用药 慎用有肾毒性的药物。
2.预防发生休克,积极抢救休克 正确处理可能引起休克的各种原发病,如已发生休克并伴有功能性急性肾功能不全时,应及时采取有效的抗休克措施,迅速恢复有效循环血量。
(二)治疗(Treatment)
1.病因学治疗 尽可能明确引起急性肾功能不全的病因,及时采取措施消除或逆转。如解除尿路梗阻、清除肾毒物、治疗肾炎等。
2.纠正水和电解质紊乱 对功能性肾功能不全应充分扩容,而对器质性肾功能不全,在少尿期应严格控制输液量,量出而入,宁少勿多,防止水中毒的发生。多尿期注意补液及钾、钠等电解质,以防脱水、低钾和低钠。
3.处理高钾血症
4.控制氮质血症 可静脉滴注葡萄糖及必需氨基酸以减轻体内蛋白质分解和促进蛋白质的很成。采用透析疗法以排出NPN。
5.纠正代谢性酸中毒
6.透析治疗 包括腹膜透析和血液透析,能有效纠正水、电解质和酸碱平衡紊乱,排出有毒物质,提高治愈率,降低死亡率,目前主张早做、多做。